Reference genetic panel analysis based on global populations and indigenous populations from the 1000 Genomes Project, Zapote and Mexican people from the Native Mexican Diversity Project
Reference genetic panels were used for various analyses of population structure. We used Global populations from the 1000 Genomes Project, Zapote individuals from Oaxaca, and Indigenous people from Mexico from the Native Mexican Diversity Project.
Maps of Mexico are created using the mxmaps R package. TheVariograms on complex traits were used to calculate distance and longitude was used to calculate latitude.
The ABCA1 variant frequencies were computed using plink in individuals from the MXB that had ancestry proxies from ADMIXTURE or had high cholesterol.
In this analysis, we removed rare variants, regions with known long-range linkage disequilibrium78,89 and variants in high linkage disequilibrium (r2 > 0.1 in a window of 50 kb and 1 variant).
The analysis we performed used a merged genotyping dataset that excluded the MAIS samples, so we could include local ancestry segments for use in the estimation of ancestry-specific allele frequencies. Before LAI analysis, reference samples were selected using the same workflow for TeraSTRUCTURE as described above, with modifications being the inclusion of 10,000 unrelated MCPS participants and an ancestry threshold of 0.95. RFMix was applied using the 753 AFR, 649 EURO and 91 AMV reference samples, a specification of 5 rounds of em and a reanalyze-reference option that treated reference haplotypes as if they were.
Genetic and environmental variables were considered to be predictors of trait variation. Polygenic scores for each trait were computed using UKB summary statistics and genetic ancestries were estimated using MAAS-MDS. The variables that we considered were latitude, longitude and altitude. We considered demographic variables of age and sex. Last, we considered sociocultural variables: educational attainment (which shows a positive correlation with income levels (Supplementary Fig. 52); however, income levels are available only for a third of the individuals); whether they speak an Indigenous language or not as a proxy for differential experience of discrimination and culture; and whether they live in an urban or rural environment. BMI was included as a covariate for all quantitative traits except height, BMI and creatinine (Fig. 5 and Supplementary Figs. 53–58 To ease interpretation of the mixed-model coefficients for jointly considered numeric and binary predictors, we standardized predictor variables as follows74. To make coefficients of numerical predictors comparable to untransformed binary predictors, we divide each variable by two times its standard deviation. We centred both the binary and numeric predictors. All of the covariates mentioned above are significant when jointly modelled for at least one tested trait, justifying their use in the full model.
The random sample of 4,000 individuals with data available for GWAS was carried out. The raw values were normalized before the analysis.
Precision biometrics of human populations in Mexico, using an ancestry based genealogical classification and a copying matrix based on Impute5
Biometric data were filtered to remove outliers with apparent errors in data entry. The height and weight of outliers was based on their distribution density over the full dataset of over 6000 individuals.
For people with blood pressure values that differed by more than 20 units for the two readings taken, the pressure was manually set and they would always have it at a normal or high pressure. Both readings were manually checked, and the readings that were not good were discarded. The updated values were used as a reference for the remaining samples. A set of adjusted blood pressure phenotypes was also generated, adjusting for treatment for hypertension. Those people receiving some form of hypertension treatment need to have 15 units added to theirs and 10 units added to theirs.
For each person, we have access to data on a wide range of factors including access to healthcare, clean water, yearly income, educational achievement, and whether or not they speak an Indigenous language.
Localities were assigned values of latitude, longitude and altitude (metres) using data from the National Institute of Statistics and Geography (INEGI) in Mexico.
Although not intended, the groupings used may seem to some as similar to racial categories that were created in the past 500 years and used to justify European superiority and colonization of global regions including present-day Mexico58,59,60. In Mexico, such categories have a similar history of racism and eugenics as in other parts of the world61. We reject fixed hierarchical categorizations of humans, as well as their use to justify the superiority of one group over another. We use ancestry proxies that we estimate from the ADMIXTURE database, and axes of ancestry that are related to these, for example, to capture variation among groups from the Americas. Genetic ancestries of humans are continuous over time and space, but should be considered only at different scales due to the complexity.
To examine fine-scale population structure using haplotype sharing, we calculated a haplotype copying matrix L using Impute5 (https://jmarchini.org/software/#impute-5) with entries Lij that are the length of sequence individual i copies from individual j. Impute5 uses a scalable imputation method that can handle very large haplotype reference panels. A Hidden Markov model is used to estimate the local haplotype sharing profile for a target haplotype and a reference set of haplotypes. The approach used to identify the subset of reference haplotypes that led to minimal loss of accuracy was based on the data structure of the PBWT. We used this method to calculate the copying matrix L, by using array haplotypes from a set of unrelated individuals as both target and reference datasets. The generated 20 PCs were created using the bigstatsr package. This is similar to the eigen-decomposition of the matrix of recipients shared segments.
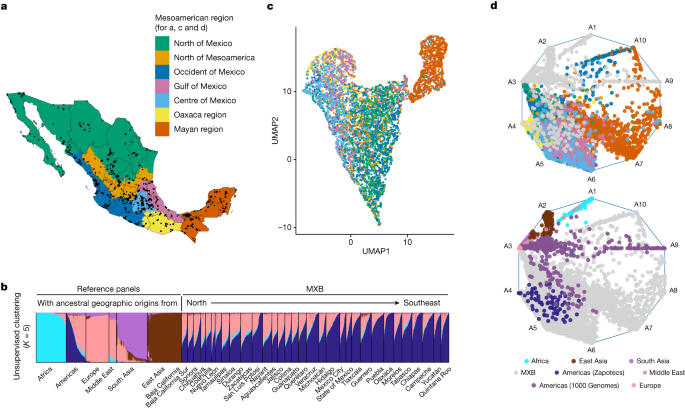
PCA and FST Analysis using smartpca in the 1000 Genomes Project Mexican Ancestry in Los Angeles, California (MXL) cohort
The smartpca was used to carry out the PCA. Principal components generated by smartpca (Supplementary Figs. 6 and 7) were used to carry out the uniform manifold approximation and projection (UMAP) analysis (Fig. 1c and Supplementary Figs. 19 and 20)63. FST analysis was done using smartpca.
This analysis was repeated in the 1000 Genomes Project Mexican Ancestry in Los Angeles, California (MXL) cohort (Supplementary Fig. 39). The effect we were seeing was due to ascertainment bias in the MEGAex array, which covers fewer rare variant, mostly native to Mexico. The whole-genome sequences from the 1000 Genomes Project allowed us to rule this out. The estimates were based on panels from the 1000 genomes, with the British in England and Scotland being the most common. Variant effect predictor was used to annotate SNPs, and mutation burden was computed in the same manner. The following consequences are included in the deleterious category.
Using AdmixtureBayes, we inferred the split events and admixture events that have occurred in the MXB. The default parameters for generating the graph were 16 and 20,000, so we used them to ensure convergence. We plotted the tree with the highest probabilities so we could see the inferred events and explore the uncertainty in the inferences. The previous use of AdmixtureBayes can be found in the corresponding paper.
Mixed-model analysis of multiple random effects for all quantitative traits: FUMA SNP2GENE and leading association SNPs and Causal groups
To assess the factors involved in creating complex trait variation, we carried out a mixed-model analysis using the lme4qtl R package for all quantitative traits. lme4qtl allows flexible model creation with multiple random effects73.
Variants were annotated as previously described38. In brief, variants were annotated using Ensembl variant effect predictor, with the most severe consequence for each variant chosen across all protein-coding transcripts. We derived annotations from a combination of APPRIS, MANE and Ensembl tags. The highest priority was given to MANE annotations. When neither MANE nor APPRIS annotation tags were available for a gene, the canonical transcript definition of Ensembl was used. The regions were defined using Ensembl release 100. Variants annotated as stop gained, start lost, splice donor, splice acceptor, stop lost or frameshift, for which the allele of interest was not the ancestral allele, were considered predicted loss-of-function variants. The missense variants that were assigned deleteriousness were SIFT, PolyPhen2 HDIV and PolyPhen2 HVAR. Missense variants were considered ‘likely deleterious’ if predicted deleterious by all five algorithms, ‘possibly deleterious’ if predicted deleterious by at least one algorithm and ‘likely benign’ if not predicted deleterious by any algorithm.
Each of the regions had been used to define lead association SNPs and potential causal groups on the basis of summary statistics for each of the associated traits. FUMA SNP2GENE was then used to identify the nearest genes to each locus on the basis of the linkage disequilibrium calculated using the 1000 Genomes EUR populations, and explore previously reported associations in the GWAS catalogue40,71 (Supplementary Table 7).
Study of how to measure the health effects of tobacco in Mexico using EDTA venous blood derived from a Buffy coat
In the 1990s, a group of British and Mexican scientists met to discuss how to measure the health effects of tobacco in Mexico. The goal was to establish a prospective cohort study that looked at the health effects of tobacco and other factors such as blood pressure, cholesterol and body mass index. Between 1998 and 2004, more than 100,000 women and 50,000 men 35 years of age or older (mean age 50 years) agreed to take part, were asked questions, had physical measurements taken, gave a blood sample and agreed to be tracked for cause-specific mortality. The study visits were mostly done during work hours when women were more likely to stay at home, and so more women were recruited than men.
At recruitment, a 10-ml venous EDTA blood sample was obtained from each participant and transferred to a central laboratory using a transport box chilled (4–10 °C) with ice packs. The samples were put in the freezer at 4 C for the rest of the night, and then moved to a dry place for 15 minutes the next morning. After being stored locally at 80 C, the samples were transported on dry ice to Oxford for further storage over liquid nitrogen. DNA was extracted from buffy coat at the UK Biocentre using Perkin Elmer Chemagic 360 systems and suspended in TE buffer. For yield and quality, samples were normalized to provide 2 g DNA at 20 ng l–1 concentration, and 2% of samples gave a minimum 1.5 g DNA.
Approximately 250 ng of total DNA was enzymatically sheared to a mean fragment size of 350 bp. Following ligation of a Y-shaped adapter, unique, asymmetric 10 bp barcodes were added to the DNA fragments with three cycles of PCR. The libraries were quantified using quantitative PCR and the s4 flow cells were fitted with Illumina NovaSeq 6000 platforms. There were a total of 10,008 samples. 200 mother– father–child trios were included. The rest of the samples are unrelated to third degree or closer, and enriched for parents of nuclear families. All samples had mean coverages over 27 and 99% of samples had average mean coverages between 3 and 38.
We separately phased the support-vector-machine-filtered WES and WGS datasets onto the array scaffold. The reference panel is made up of the phased WGS data. For the WGS phasing, we used WhatsHap (https://github.com/whatshap/whatshap) to extract phase information in the sequence reads and from the subset of available trios and pedigrees, and this information was fed into Shapeit (v.4.2.2; https://odelaneau.github.io/shapeit4) through the –use-PS 0.0001 option. A buffer was used for the beginning and end of each chunk to store the 500 SNPs from the array data. The use of the phased scaffold of array variants meant that chunks of phased sequencing data could be concatenated together to produce whole chromosome files that preserved the chromosome-wide phasing of array variants. A consequence of this process is that when a variant appeared in both the array and sequencing datasets, the data from the array dataset were used.
Similar to other recent large-scale sequencing efforts, we implemented a supervised machine-learning algorithm to discriminate between probable low-quality and high-quality variants8,12. We defined positive and negative control variants based on the following criteria: 1) concordance in the calls between array and exome-sequencing data, 2) transmitted singletons, and 3) an external set of high quality sites. The intersection of variant that passed QC in both gnomAD v.3.1 and TOP Med Freeze 8 was the first to be created. This set was additionally restricted to 1000 genomes phase 1 high-confidence SNPs from the 1000 Genomes project36 and gold-standard insertions and deletions from the 1000 Genomes project and a previous study37, both available through the GATK resource bundle (https://gatk.broadinstitute.org/hc/en-us/articles/360035890811-Resource-bundle). We put gnomAD v3.1 fail variant with topmed freeze 8 emanian or duplicate variant to define the external low quality set. Before model training, the control set of variants were binned by allele frequency and then randomly sampled such that an equal number of variants were retained in the positive and negative labels across each frequency bin. A support vector machine using a radial basis function kernel was then trained on up to 33 available site quality metrics, including, for example, the median value for allele balance in heterozygote calls and whether a variant was split from a multi-allelic site. We split the data into two categories. We performed a grid search with fivefold cross-validation on the training set to identify the hyperparameters that returned the highest accuracy during cross-validation, which were then applied to the test set to confirm accuracy. This approach identified a total of 616,027 WES and 22,784,296 WGS variants as low-quality (of which 161,707 and 104,452 were coding variants, respectively). We used a set of hard filters to exclude variant with > 10% missingness, as well as 3 mendel errors or failed Hardy–Weinberg equilibrium.
We used Shapeit (v.4.1.3; https://odelaneau.github.io/shapeit4) to phase the array dataset of 138,511 samples and 539,315 autosomal variants that passed the array QC procedure. To improve the phasing quality, we used the inferred family information to build a partial haplotype scaffold on unphased Genes, and randomly selected one offspring per family when there was more than one. We passed the scaffold information with the –scaffold option, when we ran Shapeit one by one.
Given an IBD graph Gl = (Vl, El) for a locus l, we estimated alternative allele counts and allele numbers by counting the connected components of the graph. Let the components be connected. Let CALT = {Cim: haplotypes in Cim have the ALT allele} and CREF = {Cim: haplotypes in Cim have the REF allele}
A network of IBD sharing is represented by the weight matrix, which was summed across pairs of individuals. The total length in the cM of the genome that individuals i and j share is given by each entry. We sought to create a low-dimensional visualization of the IBD network. We used a similar approach to that described in ref. The normalized graph Laplacian is used as coordinates for a low-dimensional embedded of the IBD network. If you want to see a degree matrix of the graph, you should use D. The identity matrix of the Laplacian normalized random walk graph is (L-I-D- 1W) where I am.
Source: Genotyping, sequencing and analysis of 140,000 adults from Mexico City
Phase determination of exome singleton frequencies in the Wilkinson Electron Spectroscopy System (WES) using imputed doses versus real doses at markers
We compared the imputed dosages to the true dosages at variations not on thearray to see if they were accurate. Markers were binned based on the amount of marker in the samples. The squared correlation between the concatenated vector of all the true (masked) genos at markers and thevector of all the imputed dosages at the same markers is reported in each bin. Variants that had a missing rate of 100% in the WGS dataset before phasing were removed from the imputation assessment.
$${{\rm{AC}}}{k}=\mathop{\sum }\limits{i=1}^{n}\mathop{\sum }\limits_{j=1}^{2}{p}{ijk}{G}{ij},\,{{\rm{AN}}}{k}=\mathop{\sum }\limits{i=1}^{n}\mathop{\sum }\limits_{j=1}^{2}{p}{ijk},\,{\theta }{k}=\frac{{{\rm{AC}}}{k}}{{{\rm{AN}}}{k}}$$
An estimate of the amount of population k at the site is n_k. Existing methods can be difficult to phase. The phase in which information is usedhelps to phase a portion of the singleton sites. Out of the exome singletons in WES, 45% occurred in stretches of Heterozys ancestry. For these variants, we gave equal weight to the two ancestries when estimating allele frequencies.
l: rmfor 1le jle 2 i\le N}\ {E}{l}\,=\,{({(i,j)}{l},{(s,t)}{l}):{h}^{{(i,j)}{l}}\,{\rm{and}}\,{h}^{{(s,t)}_{l}}\,{\rm{are}}\,{\rm{IBD}}}.\end{array}$$
sum _C in C_rmALTbar.
Source: Genotyping, sequencing and analysis of 140,000 adults from Mexico City
The use of Regenie to assess trans-ancestry portability of BMI PRS in MCPS and in the UK Biobank
To generate source datasets for assessing trans-ancestry portability of BMI PRS, whole genome regression was performed using Regenie (https://rgcgithub.github.io/regenie/) in individuals in the MCPS and in a predominantly European-ancestry cohort from the UK Biobank. Individuals with type 2 diabetes (ICD10 code E11 or self-reported) were excluded. The model were adjusted for age and technical co-variates to account for the fact that the models were rank based. The Regenie summary statistics from the UK Biobank were used to generate a BMI PRS in MCPS; conversely, MCPS summary statistics were applied to UK Biobank statistics.


