New development of plasmid vectors for mycoplasma using cryopreserved CD3+ cells, Lonza MycoAlert, and Codon-Optimized Gene Blocks
The coats from anonymous donors and consenting healthy donors were used in the experiments, while the human peripheral blood leukopak was obtained from the university through a board-exempt protocol. CD3+ cells were isolated using the RosetteSep Human T Cell Enrichment Kit, Lymphoprep density gradient medium and SepMate-50 tubes according to the manufacturer’s protocol (STEMCELL Technologies). There are experiments done at the Children’s Hospital of Philadelphia, and T cells from healthy donors were obtained from the University of Pennsylvania. All purified T cells were cryopreserved in CryoStor CS10 medium (STEMCELL Technologies).
The cells were transduced to express markers with or without CD19 and can be found in the table below. Single-cell clones were chosen for high expression. The 143B and Nalm6 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) and RPMI 1640, respectively, and both were supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES and 1× penicillin–streptomycin–glutamate (Gibco). Prior to the use of this study, the engineered versions of the cell lines, which were called lahm6 and 142B, were previously verified using a process called STR fingerprinting. HEK293 cells were originally obtained from the National Cancer Institute. Cells were frequently tested for mycoplasma using the Lonza MycoAlert Mycoplasma Detection kit.
The constructs used in the study are CD 19.28, CD19.BB, and anti-GD2 HA.28. Codon-optimized TCF1, FOXO1 or FOXO13A sequences and a P2A ribosomal skip sequence were generated as Gene Blocks by IDT and constructed in MSGV retroviral vectors. The only thing that the tNGFR-only construct does is not have a P2A ribosomal skip sequence. The FOXO1DBD construct was created by two steps. The plasmids were amplified by transformation and their sequence were verified by Sequencing.
American type culture collection allowed for the obtaining of retroviral packaging cell lines. The GP+E86 and tumour cell lines were maintained in the RPMI medium that was supplemented with 10% heat-inactivated fetal bovine serum. 100 U sample of penicillin and 100 g sample of streptomycin are included for 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid. The cells were kept at 37 C in a humidified incubator with 5% CO2. The PA317 cell line was maintained in DMEM (Gibco) supplemented with 2 mM glutamine and 100 U ml−1 penicillin and 100 μg ml−1 streptomycin and was maintained in a humidified incubator at 37 °C with 10% CO2.
After removing liquid nitrogen, T cells were washed with AIM-V (Gibco) supplemented with 5% FBS, 10 mM HEPES, 1 penicillin–strepomycin–glutamate and 100 U liter human IL-2. (Peprotech) or RPMI (Gibco) supplemented with 10% FBS, 10 mM HEPES, 1× penicillin–streptomycin–glutamate and 100 U ml−1 recombinant human IL-2). At a volume of 30 resuspended beads per million T cells, Human T-Expander CD3/CD28 Dynabeads were washed and added to T cells. The T cells and beads were resuspended with a concentration of 500,000 T cells per liter in T cell medium. Forty-eight and 72 hours after activation, T cells were transduced (see ‘Retroviral transduction’). Ninety-six hours after activation, beads were removed by magnetic separation using a DynaMag column (Invitrogen). The T cells were fed fresh T cell medium every 48–72 h and maintained at a density of 50 106 cells per liter after being fed. For FOXO1i experiments, T cells were provided with fresh complete T cell medium and vehicle control (dimethyl sulfoxide; DMSO) or AS1842856 (EMD Millipore) every 2–3 days from days 4 to 15 after activation.
Unless otherwise stated, tNGFR isolations were done using either MiltenyiMACS or STEMCELL EasySep. Cells were resuspended in a buffer and stained with an anti-human CD 281 (tNGFR)immunoglobulin. Cells were washed with PBS, 0.5% BSA and 2 mM After being washed with theMACS buffer and mixed with Streptavidin MicroBeads, it was passed through an LS Column for positive selection. For STEMCELL EasySep sorting, cells were isolated using the manufacturer’s protocol for the EasySep Human CD271 Positive Selection Kit II (STEMCELL Technologies) with an EasyEights EasySep Magnet (STEMCELL Technologies). After isolation, cells were immediately mixed with warm complete T cell medium, counted and resuspended at 500,000 per ml.
To interrogate the role of endogenous FOXO1 in CAR T cell function, CRISPR–Cas9 was used to delete a sequence directly upstream of the FOXO1 DNA-binding domain. On day 4 after activation, retrovirally transduced CAR T cells were removed from activation beads by magnetic separation. The reactions were prepared with a millionCAR T cells in the P3 buffer and then in the P3 primary cell kit. Ribonucleoproteins were prepared by complexing 0.15 ng of sgRNA targeting FOXO1 or AAVS1 (Synthego) with 5 µg Alt-R S.p. There is a person named Cas9 Nuclease (IDT, 1081058) before adding the cell suspension to each reaction. A previously verified sgRNA sequence was used for AAVS1 edits. For FOXO1, two separate sgRNAs were used in tandem, at equal concentrations (5′-UUGCGCGGCUGCCCCGCGAG-3′ and 5′-GAGCUUGCUGGAGGAGAGCG-3′). For TCF7 gene editing, we used a previously validated sgRNA56 (5′-UCAGGGAGUAGAAGCCAGAG-3′) for bulk RNA-seq experiments performed at CHOP. A separate sgRNA was designed and used for in-vivo experiments. The reaction was pulsed with the EH115 program on a Lonza 4D Nucleofector. Within 24 h cells were recovered immediately in 260 l of AIM-V medium, supplemented with 500 U liter IL-2 and expanded into 1 liter fresh medium. Cells were kept at a rate of up to 106 cells per liter in well plates for the duration of the study. On days 14–16, knockout efficiency was determined by intracellular transcription factor staining (Cell Signaling, 58223) followed by flow cytometry.
CAR T cells were washed twice in FACS buffer (PBS + 2% FBS) and stained with fluorophore-conjugated surface antibodies for 30 min on ice. Cells were washed again before they were analyzed. Intracellular stains were performed with the same initial surface stain, after which cells were fixed, permeabilized and stained using the FoxP3 Transcription Factor Staining Buffer Set according to the manufacturer’s protocol (eBioscience). Anti-human FOXO1 (clone C29H4) and anti-human TCF1 (C36D9) antibodies were purchased from Cell Signaling. The 1A7 anti-16g2a idiotype antibodies used for the detection of the HA CAR was derived from the NCI and was used for the Dylight 650 kit. The Dylight 650 kit was used to label the anti-FMC63 idiotype antibody. Cell-surface antibodies were used at a 1:100 dilution during staining, with the exception of anti-14g2a and anti-FMC63, which were used at a 1:1,000 dilution. Both live and dead stainings were done at a 1:1,000 and 1:50 dilutions, respectively. Cells were analysed either with FACS Diva software or with Aurora Spectro v.3.1.0. Downstream analyses were performed using Cytek SpectroFlo v.3.1.0 and FlowJo v.10.8.1 Software. All reagents are listed in Supplementary Table 2. A representative gating strategy for FOXO1KO and FOXO1OE experiments is shown in Supplementary Fig. 1. Cells were gated on all the singlets, not only dead or dying T cells, but also debris. For MFI quantification, background subtraction was performed using either unstained or FMO samples. The MFI quantification in Extended Data Fig. Background was not subtracted due to negative MFI values in some samples.
Tumour cell targets were co-cultured with CAR T cells at a 1:1 ratio for 24 h. Supernatants were collected and an equivalent amount of tumour cells were reseeded for another 24 h. This process was repeated one final time before cells were collected for analysis by flow cytometry and supernatants were analysed by cytometric bead array (CBA) using either mouse or human cytokine Flex sets (BD Biosciences) according to the manufacturer’s instructions.
The CAR T cells were prepared from different donors and the Seahorse XFe 24 Bioanalyser was used to determine the OCR. Cell culture plates were coated with l-glutamine before the cells were washed and plated onto the plates. After adherence and equilibration, cellular OCR and extracellular acidification rates (ECAR) were measured using a Seahorse MitoStress assay (Agilent), with addition of oligomycin (1 μM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (1.2 μM) and antimycin A and rotenone (0.5 μM each). 3 min mix, 3 min measurement, 3 min measurement repeat, and 3 min mix were all used in the test. As per manufacturer instruction, the values of rawOCR were normalized to the amount ofProtein per well as assessed by the Pierce BCA test. The maximum rate was used to calculate SRC.
Chromatin-bound and soluble proteins were separated as previously described23. 100 mM was used for the buffer for cytoskeletal. Aprotinin, 10 g mL, and leupeptin are 4% and 4 g, respectively. After washing with ice-cold PBS, cell pellets were lysed with CSK buffer for 20 min on ice. The sample was separated and cleared by the centrifugation at a rate of 15,870g per minute. The DC proteinASSy was used to determine the amount of thesoluble fraction’sprotein concentration. The remaining pellet containing the chromatin-bound fraction was washed twice with CSK buffer, centrifuging at 1,500g for 5 min. A boiled proteins was resuspended in a 1 Pierce reducing sample buffer. The soluble fraction was supplemented with Pierce Reducing Sample Buffer to achieve 1× and boiled for 5 min. The amounts of the two components in each sample can be analysed by SDS– Polyacrylamide gel electrophoresis and transferred to the vacumm. 30 min was allowed for the seals to be blocked in 5% milk. After washing with TBST, the cells were then placed in a cell culture medium for 7 hours at 4 C. Next, the membranes were washed with TBST and put in a petri dish for an anti-mouse or anti-rabbit experiment. IgG conjugated to horseradish peroxidase for 1 h at room temperature. Membranes were visualized using Clarity Western ECL Substrate (Bio-Rad, 1705060) and the ChemiDoc Imaging System and Image Lab Touch Software v.3.0 (Bio-Rad). After visualization, the cells were stripped with a mild stripping buffer. The previous steps were repeated for detection of soluble (1:5,000 GAPDH; Cell Signaling, 97166, clone D4C6R) and chromatin-bound (1:1,000 Lamin A; Cell Signaling, 86846, clone 133A2) fraction loading controls. The analyses were performed with the aid of the Densitometer.
Staining of tumour cells in DMEM medium with a single cell suspension and resuspension at the Peter MacCallum Cancer Centre
There were mice bred at the Peter MacCallum Cancer Centre. Nod. Cg-Prkdcscid, Il2rgtm1Wjl/SzJ, were bred at the Peter MacCallum cancer centre or obtained from Australian BioResources. Mice used in experiments were between 6 to 16 weeks of age and were housed in PC2 specific pathogen-free conditions and a minimum of 3 mice per group were used in each experiment. Each group of mice was randomly assigned to a different group based on the tumours size to ensure equal burden before therapy. Experiments were not blind as the investigators used the same techniques to analyse them. Experiments were approved by the Animal Experimentation Ethics Committee no. The ethical endpoints of all the experiments were stated in the approved projects.
Blood was collected via submandibular or retroorbital bleed into tubes containing EDTA prior to euthanasia. Blood and spleen samples were treated twice or once respectively with ACK lysis buffer before staining for flow cytometry. Tumours were digested in SAFC DMEM medium (Gibco) with 0.01 mg ml−1 DNase (Sigma Aldrich) and 1 mg ml−1 type IV collagenase (Sigma Aldrich) for 30 min at 37 °C. The samples were removed from the food and put into a 70m filter that allowed for a single-cell suspension and resuspension in the Fc block. For stimulation of intratumoral CAR T cells to assess cytokine secretion capacity, tumour cell suspensions were resuspended in complete RPMI medium with 10 ng ml−1 phorbol 12-myristate 13-acetate (Abcam), 1 μg ml−1 ionomycin (Abcam), GolgiStop (1:1,500 dilution, BD Biosciences) and GolgiPlug (1:1,000 dilution, BD Biosciences). When the samples were in 37 C with 5% CO2, they were ready to be stained for analysis. Single-cell suspensions from dLN were created by placing tissue between two pieces of 70-μm filter mesh in 400 μl of FACS buffer and by mechanically digesting using the end of a syringe. The resulting cell suspension was stained for analysis. According to the manufacturer’s protocols, isolated cells were stained using MitoTracker Deep Red and Mitotracker Green.
The T cells were pelleted usingcentrifugation and flash-frozen. The pellet was processed using a mini Kit or an AllPrep DNA/RNA Micro Kit, according to the manufacturer’s instructions. A 150 bp read length and around 50 million read pairs per sample was used to quantitate total RNA with a Qubit Fluorometer.
The FGsea package was used to perform an unbiased enrichment analysis on genes that were pre-ranked by fold change. There are referencegene sets obtained from the MsigDB library as well as CHEA dataset or analyses of scRNA.
Cell selection was used to generate single-cellRNA-seq libraries of CAR T cells and Her2+ tumours were collected from five mice per condition. Tumour-infiltrating CAR T cells were further purified by sorting human CD3+ TILs from each isolate using a Cytek Aurora Cell Sorter. A total of 20,000 CAR TILs were sorted from each tumour and pooled across five mice per group. Cells were barcoded and sequencing libraries were generated using the 10X Chromium Next GEM Single Cell 3’ v.3.1 kit (10X Genomics) according to the manufacturer’s instructions. Libraries were sequenced at the CHOP High Throughput Sequencing Core on an Illumina NovaSeq 6000 with an average read depth of 50,000 reads per cell.
The pepatac library was processed using the default options. Fastq files were pre-aligned to theMitochondrial genomes to exclude reads from them. To make sure the data was accurate, multiple reads aligning to repetitive regions of the genome were removed from the data. Bowtie2 was then used to align the reads to the hg38 genome. SAMtool was used to identify uniquely aligned reads and Picard was used to remove duplicate reads. The resulting deduplicated and aligned BAM file was used for downstream analysis. Peaks in individual samples were identified using MACS2 and compiled into a non-overlapping 500-bp consensus peak set. The peaks were scaled down to 500 bp width and ranked by significance. The most significant peak was retained even though the peaks that overlap with the region were selected by ranks. The peak-sample count matrix was generated using ChrAccR with the default parameters of the run_atac function. Signal tracks for individual samples were generated within the pepatac pipeline. The information from the tracks was merged with that from all the samples to produce a comprehensive view.
The peak-sample count matrix has a threshold of an absolute log 2-transformed fold change greater than 0.2 and P values less than 0.10 being used to identify differential peaks. There were adjusted P values that weren’t used. The vst function is what we used to get the variance-stabilized count matrix. We took effect from donor using the removeEffectBatch function in the limma library. Finally, we generated PCA plots using the corrected matrix with the plotPCA function using the top 2,000 most variable peaks. We aggregated differential peaks across conditions, standardized the peak signals using z-scores across samples and performed k-means clustering to generate a chromatin accessibility heat map. Motif enrichments of differential peaks and grouped peaks were searched with HOMER and findMotifsGenome.pl with default parameters. The gchromVAR package was used to enrichment the regulatory elements. The method weights the chromatin features by the log2transformed fold changes from a previous report and uses the data to calculate the enrichment for each cell type.
The FOXO1 regulon gene set was created with intersecting downregulated differential genes and up regulated differential genes. The regulon enrichment scores were calculated using ss GSEA in the GSVA R package.
For regulon analyses of single-cell ATAC-seq data, the processed Signac data objects of CAR T products profiled in a previous study were obtained. For consistency between donors, the mean fragments in peaks per cell were down sampled. Due to the high data quality, the donors were excluded from being included in the study. We computed the per-cell epigenetic signature per factor using the chrom VAR workspace and related T cell signatures derived from bulk experiments. In order to compare the associations between responders and non- responders with this signature, we did an ordinary least squares regression with the per-cell Z-score. The Wald test statistic was used to calculate the significance of the two regressions for each factor.
Unless otherwise stated, statistical analyses for significant differences between groups were conducted using one- or two-way analysis of variance (ANOVA) with Bonferroni, Tukey’s or Dunnett’s multiple comparisons test, or with a Student’s or Welch’s t-test using GraphPad Prism v.9.4.1. We used a student’s t-test in one of the experiments in which same-donor samples were compared. Survival curves were compared using the log-rank Mantel–Cox test. Statistical methods were not used to predetermine sample sizes.
The protocols of this research have been approved and are in compliance with Peter MacCallum’s ethical guidelines regarding using animals. Studies using human peripheral blood mononuclear cells (PBMCs) from healthy donors was approved by the Peter MacCallum Cancer Centre Human Research Ethics committee. Informed consent was obtained from the Australian Red Cross.
The isotypecontrol (2A3 clone, IgG2a, BE0254) and Mouse IFN antibody were purchased from BioXcell. The cytokine IL-2 was obtained from the National Institutes of Health and purchased from Peprotech. IL-7 Peprotech bought IL-15. CAR T cells were stimulated with an anti-idiotype antibody that was custom made.
Mouse TCF7, FoxO1, ID3 and JUNC were cloned into the mouse stem cell viruses, which are either an mCherry marker Gene or truncated (lacks cell signalling components). The viral packaging GP+E-86 cell line that produces the anti-HER2 CAR retrovirus was generated as previously described48. The anti-HER2 CAR was made with an extracellular scFv specific for human HER2 and an CD8 hinge region, a CD28 transmembrane domain and an intracellular CD3 domain. The GP+E-86 cell lines were used to generate anti-HER2 CAR cells and they were sorted based on their NGFR or mCherry expression. CAR T Cells were maintained in RPM supplementedI medium with -mercaptoethanol and IL-7 as a precautionary measure, after the cells were transduce primary mouse T cells.
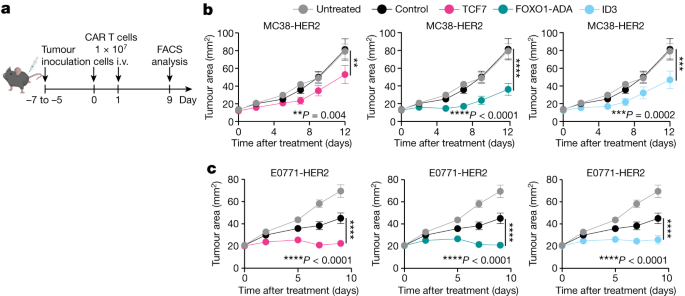
In C57BL/6 mice, 2 105 E0691-HER2 breastcancer cells were injected into amammary fat pad 5 days prior to treatment or 2.5 105 MC38-HER2 colon adenocarcinoma cells 5 days prior. After tumours were established, mice bearing E0771-HER2 or MC38-HER2 tumours were preconditioned with 4 Gy or 0.5 Gy total body irradiation respectively. Mice were then treated with intravenous doses of 1 × 107 CAR T cells on 2 consecutive days and one dose of IL-2 (50,000 IU per dose) with the first dose of CAR T cells, followed by two doses of IL-2 each day on the next 2 consecutive days. Tumour area was measured every 2–3 days following treatment. The mice were given 300 g of anti-IFN or isotype control antibodies on the first three days after CAR T cell treatment.
GraphPad Prism for Analysing Multiple Datasets and Groups of Data (Content Algorithm and ANOVA)
Statistical analyses were performed using GraphPad Prism. Analyses are performed to compare two datasets, one-way ANOVA to analyse multiple datasets across a single time point and two-way ANOVA when analyzing several sets of data across time.