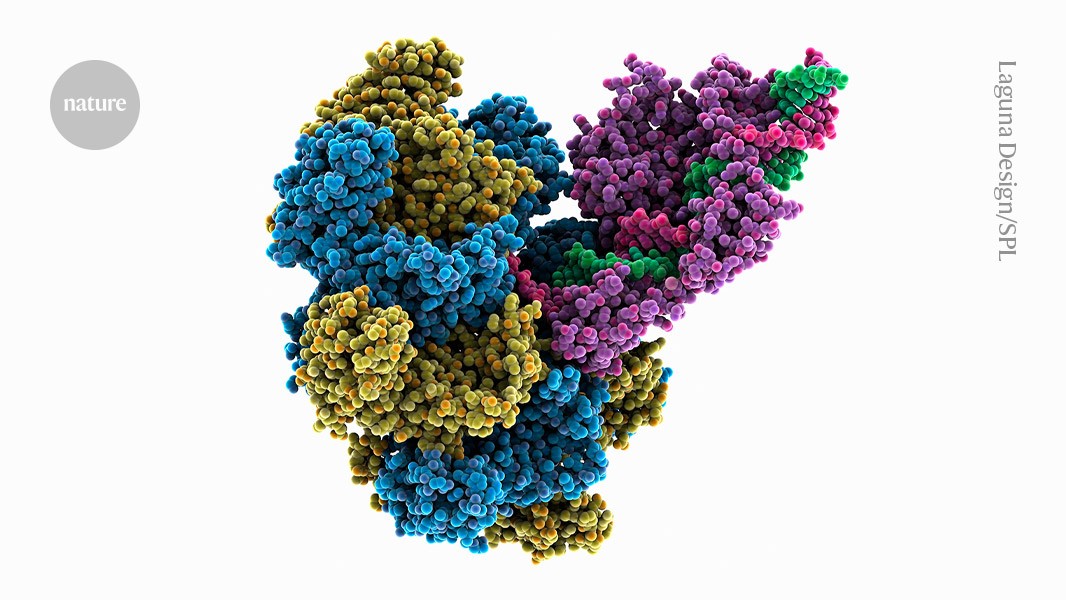
Introducing full-length genes via a CRISPR-associated transposase: How efficient was evoCAST?
The system circumvents both of these problems by introducing full-length genes at targeted sites in a single step. Developed by Liu, biochemist Samuel Sternberg at Columbia University in New York City and their colleagues, the method uses a bacterial enzyme complex called a CRISPR-associated transposase, or CAST.
Described today in Science1, the method could pave the way for gene-correction therapies that would be given once, and work regardless of the specific mutation causing an individual’s disease. It could also accelerate the development of engineered cell therapies for cancer and simplify the creation of genetic models for research.
“It could really be a big part of the future,” says study co-author David Liu, a chemical biologist at the Broad Institute in Cambridge, Massachusetts.
Transposases are enzymes that power the movement of ‘jumping genes’ — ‘selfish’ bits of DNA that hop around the genome to propagate themselves. The CAST systems were already used to shuffle large pieces of genetic material. In human and other mammals, the previously reported versions of CAST struggled with low efficiency.
After hundreds of viral generations, and rational engineering of some CAST components, the researchers produced an optimized version of the enzyme complex. The achievement pushes the boundaries of design due to 21 small changes across five proteins, says a bio engineer at Riken in Wak, Japan. This is crazy directed evolution. he says.
The resulting complex, termed evoCAST, demonstrated an insertion efficiency of up to 30% across multiple genomic sites — a more than 400-fold improvement over the non-evolved original.
The Muldoons told reporters this week that their baby is doing well after receiving three doses of a treatment that was supposed to fix a genetic flaw. But it is too soon to use the word “cure”, says Rebecca Ahrens-Nicklas, a paediatrician at Children’s Hospital of Philadelphia in Pennsylvania, and one of Muldoon’s physicians. “This is still really early days,” she says. We know we have more to learn from him.
In six months, a group of clinicians and researchers in both industry and academia began to develop Muldoon’s therapy. The drug was specific to Muldoon’s genetic sequence, and won’t ever be used for another person.
He did not produce the normal form of the bread-and- butteridase, dubbedCPS1, because he had two different mutations from each of his parents. This compromised his ability to process the nitrogen-containing compounds produced when the body breaks down protein. As a result, his blood had high levels of ammonia, a compound that is particularly toxic to the brain.
Muldoon wouldn’t be eligible for a liver transplant for months. Only half of babies with severeCPS-1 deficiency who survive long enough to receive a transplant survive the added risk of brain damage or death each day.


