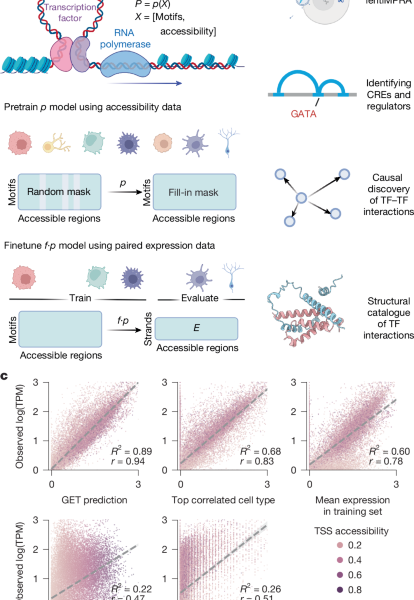
Modeling and stratification of the large-scale single-cell accessibility data for K562 (lentiMPRA) using a tetrahedral gene model
We trained for the large-scale single-cell accessibility data. The model on the pair of access–gene expression data has the same log-likelihood loss function as Enformer4, and we fine-rigged it.
To simulate this approach using GET, we first collected the sequence element library and constructed the vector sequence for insertion, including both the regulatory sequences and mini promoters. We then followed the same data preprocessing procedure to get the motif scores of the inserted elements. For each element, we performed in silico insertion by summing its motif score with an existing region on the genome. The sample of 100 regions was then used to make prediction of what would happen. The mean predicted expression was compared with the mean predicted accessibility as a prediction of regulatory activity. For each region, we performed 600 insertions across the genome to match the experimental insertion count. We used the model that was fine-tuned to perform the inference. 5 days is how long it took to complete all 200,000 elements in K562.
We stratified the K562 lentiMPRA elements (approximately 200,000) by overlapping the annotated 15 ENCODE ChromHMM states computed from histone mark and other ChIP–seq data for K562. We selected the elements overlapping with states ‘12 EnhBiv’, ‘6 EnhG’ and ‘7 Enh’ as enhancers, and those overlapping with ‘13 ReprPC’, ‘14 ReprPCWk’ and ‘15 Quies’ as repressive and quiescent regions.
Genome-by-Motif: Learning the Jacobian Tensor for a Cell Type-Specific ATAC Model
The Jacobian matrix (tensor) JX ∈ ({{\mathbb{R}}}^{r\times 2\times r\times m}) of (f) at the point (E, X) evaluates how each output dimension will change when each input dimension changes by a small quantity. We specifically pick the output dimension and strand that correspond to the given gene, represented by ({\rm{\nabla }}g\in {{\mathbb{R}}}^{r\times m}):
The feature (motif) importance vector ({{\rm{v}}}_{g}\in {{\mathbb{R}}}^{m}) is obtained by multiplying the gradient element-wise with the original input and summarizing across regions:
The Hadamard product is what (odot ) means. Even though the (X) has a quantitative ATAC signal, we still use it to infer (J_X) in abinary ATAC model. This facilitates study of the relationship between regulators and observed chromatin accessibility.
The cell-type-specific genome-wide Gene-by-‘motif matrix for cell types c,Vc is acquired by concatenating the. The same process can be applied to different cell types.
We used multiple feature attribution methods in different analyses and provided all options to users in our packages. More specifically, the gradient of the model’s output with respect to the input features, represented by the vector (\nabla f(x)), measures how much the model output (expression) will change when we change a small amount of the input along a dimension (for instance, a certain motif in a cis-regulatory region). In the context of neural network features, the generalization is made to multiple outputs of the Jacobian matrix. It’s important to understand the influences of individual features on the transcription levels in order to understand the Jacobian matrix.
We can use the gene-by-motif matrix to find out which genes will be most affected by a TF by using the largest entries in the motif column. We chose the top 1000 genes and did a gene enrichment analysis using g:Profiler. We used the term size more than 500 and less than 1,000 to sort the results. Terms with adjusted P value less than 0.05 were retained as significant terms. We further selected TFs in the ‘Hemopoiesis’ term with expression log10(TPM > 1) for visualization against the GATA motif score.
The input matrix was collected from the hepatocytes or from any fetal or adult cell type. Every pair of motifs received a score for Pairwise Pearson correlation. We performed the discovery for all cell types using the gene-by-measures matrix for the cell type specific motifs. Interactions with the top 5% absolute effect size were retained in the final database. For each interaction, we performed structural analysis between the two TFs with the highest expression in the corresponding cell types.
GET is designed to capture the relationship between regulators and regions. We needed every input sample to have a certain number of consecutive accessible regions in order to facilitate this. Through previous experiments, we found that the ideal equivalent genome coverage is around or more than 2 Mbp, a range in which most of the chromatin contact happens. We chose 200 input regions per training sample based on our current data. Pretraining and sliding window approach were used. The sliding window’s stride had to be reduced to match the number of regions in one sample.
We used the cell-type-specific and cell-type-agnostic settings to perform Spearman correlation. Input × gradient scores were used to construct the matrix for computational efficiency. For the cell-type-specific settings, all genes with promoter overlap with open chromatin peaks in each cell type were used in the correlation calculation. Causal discovery was done using the LiNGAM69. For the cell-type-agnostic settings, 50,000 genes were randomly sampled from all cell types, and the resulting matrix was subjected to the LiNGAM algorithm implemented in the Causal Discovery Toolbox Python package with default parameters.
To calculate the motif binding score within a specific genomic region, the corresponding sequence was scanned against the hg38 reference genome, using 2,179 TF motif position weight matrices previously compiled by Vierstra et al.13 (accessible at https://www.vierstra.org/resources/motif_clustering). The threshold 61 was used for the scanning process with the MOODS tool.
pLDDT is a reliable caller of the Protein domain owing to its accurate structure prediction performance. Each sequence was categorized into two high and low pLDDT regions. Empirically, we found that 80% (recall) of known DNA-binding domains could be easily identified using high pLDDT regions plus a high ratio of positively charged residues. We normalized the score by dividing it by the maximum and first computed smoothed pLDDT using a10-acid moving average. The region with a smoothed pLDDT score less than 0.4 was defined as a low pLDDT region. If two low pLDDT regions were close (less than 30 amino acids), they were merged into one. Any region that was not a low pLDDT region was labelled as a high pLDDT region.
LocalColabFold and ColabFold were used to predict multimer structures with the AlphaFold Multimer v.2.3 model. For homodimer prediction, we used all five models with three recycles. The model 3 we used for the large-scale interaction screening was used with three recycles. The pAE and pLDDT were stored for downstream analysis. pDockQ was calculated using the code FoldDock70.
Source: A foundation model of transcription across human cell types
Proximity labelling of HeLa and REH B-ALL cell lines for coimmunoprecipitation negative controls
He La cells and REH cells were bought from ATCC. Cell lines purchased from a certificated bank weren’t further authenticated. All cell lines tested negative for mycoplasma. The study did not use commonly mis identified cell lines.
The cells were cultured in a lab. supplemented with 10% defined fetal bovine serum (HyClone, SH30070), at 37 °C and 5% CO2. HeLa cell lysates were created with a lysis buffer of 50 mM tris-hcl, 150 mM NaCl, and NP-40. The samples were first put to bed with 5 g agarose-conjugated TFAP2A primary antidote before being run in a Laemmli loading buffer. The separated Tris–glycine gels were transferred to the iwmmobilon-P, which then probed with primary antibodies against TFAP2A. A repeat experiment was performed for coimmunoprecipitation negative controls, which were probed with primary antibodies against SRF (Abclonal, A16718, 1:750) and β-actin (Cell Signaling Technology, 4967, 1:10000), followed by chemiluminescence detection.
We initially cloned PAX5-WT and the PAX5 G183S mutant into the pCDNA3.1-MCS-13Xlinker-BioID2-HA (Addgene, catalogue no. 80899)71. Wecloned PAX5-WT-13Xlinker-BioID6-HA and Pax5-G183S13Xlinker-bioID8-HA to the pCDH-GFP-puro vector. We used pCDH-PAX5-WT-13Xlinker-BioID and pCDH-G183S-13Xlinker-BioID to transduc the REH B-ALL cell line. The proximity labelling assay was performed following previously published methods71,72,73. Briefly, REH stable cell lines with control vector pCDH-13Xlinker-BioID2-HA-GFP, pCDH-PAX5-WT-13Xlinker-BioID2-HA-GFP and pCDH-PAX5-G183S-13Xlinker-BioID2-HA-GFP were incubated with 100 μM biotin (Sigma-Aldrich, B4501) for 24 h. We collected the cells, washed them twice in cold phosphate-buffered saline and incubated them for 50 min on ice with occasional vortexing in lysis buffer (150 mM NaCl, 10 mM KCl, 10 mM Tris-HCl pH 8.0, 1.5 mM MgCl, 0.5% IGEPAL) supplemented with protease and phosphatase inhibitors (Life Technologies, catalogue no. 78443) and 63 U of benzonase (Sigma-Aldrich, catalogue no. 70746-3). Proteins were clarified by centrifugation at 21,000g for 15 min at 4 °C. We performed total protein quantification using a Pierce BCA Protein Assay kit (ThermoFisher Scientific, catalogue no. 23225) and incubated 1 mg of total protein extract with 100 μl of magnetic streptavidin beads (Dynabeads MyOne Streptavidin C1, Life Technologies, catalogue no. 65002) on a rotator at 4 °C overnight to isolate biotinylated proteins. The beads were washed twice with lysis buffer and once with 2 M urea. Twice before with lysis buffer the Tris-HCL was pH 8.0. Biotinylated proteins were eluted by boiling in 4× protein loading buffer supplemented with 2 mM biotin and 50 mM dithiothreitol at 95 °C for 10 min. The western blotting procedure can detect lysosomal riboflavin with the following antibodies and protocols: Standard protocols and Strepsavidin– HRP. Anti-PAX5 is one of the anti-HA items in the Cell Signaling catalogue. Proteins were detected using a Li-Cor Odyssey OFC instrument and quantified using the GelAnalyzer 23.1 software.
The experimental procedure involves designing alibrary of lentiviruses that contain desired sequence elements and a mini promoter. The regulatory activity is measured through the use of genomic data as well as the log copy number of the RNA and integrated DNA.
Source: A foundation model of transcription across human cell types
3d Booster-Gene Pair Recognition Using a Simple Two-Dimensional Neural Network with Distance Contact Map
It is possible that the model could be changed in future work by taking GET region embeddings as further input and learning to predict three-dimensional contacts.
All scores in this benchmark (ABC, Enformer, GET, HyenaDNA, DeepSEA and DNase/ATAC) were further normalized across each gene’s ±100 peaks to make them comparable across genes.
Recent studies have highlighted the dominant importance of one-dimensional genomic distance in governing CRISPRi enhancer knockout effects (for instance, Gschwind et al.33). In this benchmark, most methods include a component of genomic distance. Enformer has exponential decay in its encodes. The results of our benchmarking are tied to the exponential decay from the TSS as shown in Figure 3c. We have also extended GET to incorporate distance information. We designed a simple module for getting a pairwise one-dimensional distance map from peaks to a pseudo-Hi-C map. The distance contact map is a simple two-dimensional neural network with a log and a SCALE-normalized observed contact Frequency as input. The model was trained by using a negative log-likelihood loss. A 0.855 Pearson correlation was captured for training ABC Powerlaw and DistanceContactMap, which were both trained with the same K562 Hi-C data. We termed the prediction of this model ‘GET Powerlaw’. There are two more scores shown in the picture. 3d are defined as follows:
We used a model that was the biggest pretrained model on the market. To score enhancer–gene pairs, we performed in silico mutagenesis by knocking down the enhancer element (that is, setting each base pair in the enhancer region to the unknown nucleotide N in the vocabulary set) and comparing against the wild-type likelihood of observing the promoter sequence.
Enformer: we used Enformer’s contribution score (gradient × input) with background normalization, following the normalization procedure described by Gschwind et al.33.
Source: A foundation model of transcription across human cell types
Cross-cell-type prediction beyond astrocytes using ABC Powerlaw and K562 Hi-C: data analysis and library size constraints
ABC Powerlaw is computed based on the powerlaw function in the official ABC repo, along with the data from K562 Hi-C and values provided in the same repo.
The main model used in our analysis was the ATAC model. This approach ensures that the model doesn’t rely on accessibility signal strength as a surrogate for sequence characteristics.
There are certain characteristics of cellular architecture that can contribute to certain variations in model performance. We show that GET can be applied to non-physiological cell types as well as capturing cell-type specific information. Beyond the intrinsic biological differences between cell types, we believe the following factors could also affect performance when generalizing to new datasets.
We validated the cross-cell-type prediction performance beyond astrocytes to include a broader range of cell types. Fetal cell types had a variable length peak set that was used for the benchmark. This comparison includes a data set of data from the Basenji15 training set and inferred genes from the Basenji test set, as well as a training cell type mean expression baseline. We used Pearson correlation, Spearman correlation and R2 to evaluate prediction performance in all settings.
Cell type rarity and library size: rare cell types often have smaller data libraries, which can limit the model’s learning potential and affect the accuracy of predictions.
Source: A foundation model of transcription across human cell types
A multioutput linear regression model for the leave-one-chromosome-out Benchmark across fetal and adult tumour cells from the Human Tumor Atlas Network
Logistic regression is a good choice for linear regression since our setting is better aligned with regression than classification. The scikit-Learn LinearRegression and MultiOutputRegressor were used.
We performed a leave-one-chromosome-out Benchmark across all chromosomes and found that the performance remained consistent as long as the platform and data sources were the same. We found an average Pearson correlation of 0.78 (minimum: 0.73, maximum: 0.84) on fetal astrocytes. We also extended our evaluation of leave-out chromosomes to tumour cells from patients with IDH1 wild-type GBM from the Human Tumor Atlas Network. We performed tuning of the base model on a single patient and looked at performance on each leave-out chromosome. This evaluation showed an average Pearson correlation of 0.75 (minimum: 0.68, maximum: 0.81) on leave-out chromosomes.
The base model for Qatac was trained on the fetal and adult Atlases with binarized ATAC signals. In the fine-tuning, we used the original aCPM for ATAC signal.
QATAC from QATAC fine-tuned: in this setting, the base model was the leave-out-astrocyte RNA-seq prediction model trained on the fetal accessibility and expression atlas. We further fine-tuned this model using quantitative ATAC signal.
Source: A foundation model of transcription across human cell types
Adapting GET to functional genomic assays using Tn5 alignment counts and chromosomes in K562 bulk ATAC data
Significant gains in time and storage complexity can be achieved thanks to these experiments. On a single RTX 3090 GPU, all fine-tuning converged within 30 min, resulting in a 3 MB K562-CAGE-specific adaptor that could be merged into the base model.
Here are the results of transfer of pretrained GET to different functional Genomics assays. We collected the data for K 562 bulk ATAC prediction. After calling peaks using MACS2 with default parameters, we computed the log(aCPM) by counting Tn5 insertions located inside the peak and filtered out the peaks with log(aCPM) less than 0.03. The corresponding peaks were used for prediction and scanning. 200 peaks per input sample was used for leave-one-chromatosome-out fine-tuning. The base checkpoint was trained on the fetal and adult atlas with a binarized APAK setting and 200 peaks per sample. LoRA was used for all layers. Each fine-tuning took around 160 s to complete eight epochs, after which the model started to overfit. Pearson correlation was collected at eight epochs for all fine-tuning. We obtained the K562 CAGE file from FANTOM5 and used bed tools to get alignment counts in peaks from ENcode K562 scATAC-seq data. 200 peaks per sample was used for fine-tuning, depending on how ATAC information was used in conjunction with features.
When leaving out one to ten motifs, GET showed a robust performance. The performance was degraded heavily when using 20 motifs with a top 20% cutoff for each motif independently, owing to removal of most of the training data.
Owing to these biases, it is difficult to directly apply a model trained on one dataset to a new platform without fine-tuning. We used a leave-out cell type approach to fine- tune for the new dataset. For a dataset of sorted cell types where only one cell type was available, we used leave-out chromosome training.
The main challenge in adapting the model to the new data is to make sure that input spaces are compatible. ATAC peak sets can be vastly different due to variation in cell types, technologies and preprocessing lines. To create a compatible peak set, we combined new and training peak sets. When overlaps occur between peaks we assign priority to the training peak set coordinates. Unique peaks from the new data are incorporated as they are. We used a uniform peak calling pipeline to maintain consistent peak lengths (for instance, 400 bp in the fetal–adult atlas) across training and new datasets. The total peaks of the fetal-only/fetal-adult set are usually less than 10% of the total peaks. This approach has demonstrated promising transferability to various data types, including SHARE-seq data of perturbed human embryonic stem cells and 10× multiome GBM data.
Source: A foundation model of transcription across human cell types
Self-supervised Training in Scintillator Random Forest: An Introduction to the Implementation and Scaling Criteria of Self-supervised Training
Random forest: we used scikit-learn RandomForestRegressor with ten estimators and max depth 10. Two-dimensional output was handled by MultiOutputRegressor.
CNN has three layers, 283 input, 128, 64, 32, 3Kernel size, followed by FC, softplus and ReLU, to give you an idea of the layers. The same parameters were used in the optimizer and for the scheduler as in theGET.
GET provides an option for fine tuning over any specific layer. This is commonly used to adapt to a new assay or platform; we apply LoRA to the region embedding and encoder layers, while doing full fine-tuning on the prediction head. This markedly reduces 99% of the parameters.
The use of early stopping enabled us to pick out the best model checkpoint for subsequent evaluation.
Where (rmz_lprime, rmz_l-1) represents the intermediate representation in the block. The feed-forward network layer has two linear layers with a GELU activation layer.
The regions that were selected were replaced with a token which is a shared but learnable token. The training goal is to predict the original values of the masked elements (M). We take a regulatory element as input to get, and a linear layer as the prediction head. Therefore, the overall objective of self-supervised training can be formulated as:
The PyTorch framework is used for the GET implementation. AdamW was applied as our optimizer in the first stage and had a weight decay of 0.05 and a batches size of 258. The model was trained to scale in a linear fashion. We set the maximum learning rate to 1.5 × 10−4. It typically takes around a week for the training to be done. For the second fine-tuning stage, we used AdamW63 as our optimizer with a weight decay of 0.05 and a batch size of 256. A model is trained for 100 epochs and then completed in eight hours using eight A 100 computers. Inference for all genes in a single cell type takes several minutes, making it possible to perform large-scale screening.
We include a more detailed description of the optimization hyperparameters, computation infrastructure and convergence criteria used in the development of the model in the section below.
The pretraining phase is similar to the Computational Infrastructure, where fine- tuning was done on eight NVIDIA A 100 GPUs.
Epochs and duration: the fine-tuning process was shorter, consisting of 100 epochs, and completed in around one day. This phase was critical for tailoring the pretrained model to specific tasks.
Source: A foundation model of transcription across human cell types
Mapping Accessibility and Expression in Multiome Data Using Peak Calling and Log10(TPM + 1) Measurements of Detection and Filtering
$${{\rm{z}}}{l}^{{\prime} }=\text{MHA}(\text{LN}({{\rm{z}}}{l-1}))+{{\rm{z}}}{l-1}\,;{{\rm{z}}}{l}=\text{FFN}(\text{LN}({{\rm{z}}}{l}^{{\prime} })) +{{\rm{z}}}{l}^{{\prime} },$$
where ({W}{q},{W}{k}\in {{\mathbb{R}}}^{(n\times D)\times {d}{{\rm{k}}}},{W}{v}\in {{\mathbb{R}}}^{(n\times D)\times {d}_{{\rm{v}}}}) are learnable linear transformations.
For identification of cell-type-specific accessible regions, the peak calling results from the original studies of each dataset were used to obtain a union set of peaks. Subsequently, to compile a list of accessible regions specific to each cell type, we filtered out peaks with no counts.
The count of fragments within a given region for a particular cell type pseudobulk is used to calculate the accessibility score for a specific genomic region. The log CPM procedure normalized the counts so that they could better be used in the model. The total fragment count in a pseudobulk and the fragment count in region i can be used to calculate the accessibility score.
For experiments encompassing multiomics, the correspondence between accessibility and expression was inherently determined through cell barcodes. In pseudobulk cases, where accessibility and expression were assessed independently, cell type annotations were used to facilitate the mapping. Specifically, the fetal expression atlas from Cao et al.23 was used for fetal cell types, whereas adult data were extracted from Tabula Sapiens24. When several ATAC pseudobulk shared the same cell type annotation, identical expression labels were assigned. The current lack of multiome data is expected to change dramatically in the future and necessitated this compromise.
We used log10(TPM + 1) to improve training stability. To overcome the problem of most scRNA-seq quantification being at gene level, not transcript level, we mapped the gene expression to accessible regions using the following approach: if a region overlapped with a gene’s TSS, the gene’s expression value was assigned to that region as a label; if a region overlapped with multiple genes’ TSS, the expression values of the corresponding genes were summed, and the sum was used as the label of that region; if a region did not overlap with any TSS, the corresponding expression label was set to 0. Further, if a promoter had very low accessibility (for instance, an accessibility CPM (aCPM) less than 0.05), we also set the corresponding expression value to 0. The regulatory element was assigned to an expression target value.
In alignment with the 200 × 283 input matrix, the target input is a 200 × 2 matrix, symbolizing the transcription levels of the corresponding 200 regions across both positive and negative strands.