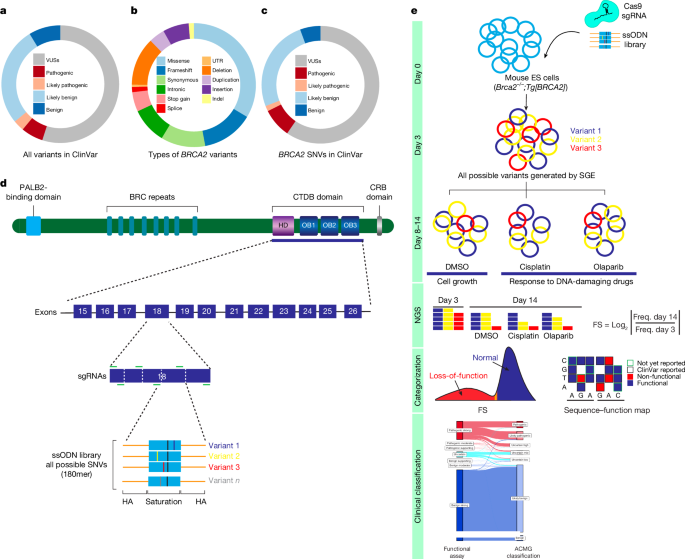
There is a functional evaluation and clinical classification of the BRCA 2 variant
Deep Learning Prediction of Genome-Wide Variant Effects: CADD-Splice-Improving Prediction Using Deep Learning-Deduced Splice Scores
Rentzsch, P., Schubach, M., Shendure, J. & Kircher, M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 13, 31 (2021).
Hu, C. There are 752 germline BRCA2 missense variant affecting the DNA binding domain. Am. J. Hum. Genet. 111, 584–593 (2024).
Source: Saturation genome editing-based clinical classification of BRCA2 variants
An automated bioinformatics pipeline for in silico Saturation Mutagenesis: the case of the protease and the bbac074 samples
Tiberti, M. et al. A program for in silico saturation mutagenesis of theProtein structures. Brief. The bbac074 is a Bioinformatics 23.
de Bruijn and I. Analysis and visualization of longitudinal genomic and clinical data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 83, 3861–3867 (2023).
Sorrentino, E. et al. In an existing bioinformatic pipelines, VarSome is integrated for automated ACMG interpretation of clinical variants. Eur. Rev. Med. It is the science of medicine. The article is titled ‘Sci. 25, 1–16 ( 2016)’.
Walker, L. C. Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: recommendations from the ClinGen SVI Splicing Subgroup. Am. Someone called it J. Hum. Genet. Over 20 years, with the number-110.
A computational model for classification of BRCA2 variants based on saturation genomic editing-based clinical classification by multiplex homology-directed repair
Breast cancer and ovarian cancer cases and associated clinical phenotypes were collected from individuals receiving cancer genetic testing by Ambry Genetics. Publicly available reference controls were women from gnomAD (v.2.1, v.3.1 and v.4 excluding the UK Biobank). The UKBiobank.ac.uk provided matching case–control data for breast cancer as well as population-based breast cancer studies and case– control data from CARriERS and BRIDGES. The analyses did not includeVariants with an allele frequency of >0.001.
There are needles in stacks of needles, and they are being used to find disease-causal variations. Nat. Rev. Genet. 12, 628–640 (2011).
Tabet, D., Parikh, V., Mali, P., Roth, F. P. & Claussnitzer, M. Scalable functional assays for the interpretation of human genetic variation. Annu. Rev. Genet. It was 56 on 19.1–19.25.
Findlay, G. M., Boyle, E. A., Hause, R. J., Klein, J. C. & Shendure, J. Saturation editing of genomic regions by multiplex homology-directed repair. Nature 513, 120–123 (2014).
The book was written by K. et al. A computational model for classification of BRCA2 variants using mouse embryonic stem cell-based functional assays. There is a Genom called NPJ. There are 52 Med. 5, 52 in 2020.
There are cancer genes and there are different types of cancer genes. In the next few years, Genome Biol. 22, 80.
Source: Saturation genome editing-based clinical classification of BRCA2 variants
Analysis of the BRCA2-DSS1 interaction in a cell-based assay for the assessment of the PS3/BS3 criterion using CountReads
A P., and Mishra were involved in the project. BRCA2–DSS1 interaction is dispensable for RAD51 recruitment at replication-induced and meiotic DNA double strand breaks. Nat. Commun. 13, 1751 (2022).
Tavtigian, S. V., Harrison, S. M., Boucher, K. M. & Biesecker, L. G. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. It’s a sad thing. It was Mutat. 41, 1734–1737 (2020).
Brnich, S. E. et al. Recommendations for use of the ACMG/AMP sequence variant interpretation framework for the PS3/BS3 criterion. The journal Genome Med. published a report on the 3rd of September.
K. et al. A comprehensive functional characterization of BRCA2 variants associated with Fanconi anemia using mouse ES cell-based assay. In 2430 there is Blood 118.
Arnaudi, M. et al. MAVISp: multi-layered assessment of variants by structure for proteins. Preprint at bioRxiv https://doi.org/10.1101/2022.10.22.513328 (2023).
FASTQ files of sequenced samples from Illumina MiSeq or NextSeq assays were trimmed for adapter sequences using cutadapt (v.3.5). SeqPrep (v.1.2) converted the paired-end reads into single reads. The single reads were aligned to the human reference genome (GRCh38) utilizing bwa-mem (v.0.7.17). After alignment, a custom developed tool called CountReads was used to analyze the data in a particular way. CountReads included the preparation of reference amino acid and DNA sequences, validation of sequencing data integrity and precise trimming of reads to relevant regions. The method confirmed that there were specific variations and aggregated and reported variant data. CountReads produced a variant call format (VCF) file, which was annotated using CAVA35. The tools were utilized to assess the effects of all the observed SNVs.
The log2 ratio between the frequency of D14 and D0 read counts was used to measure the depletion or enrichment effect for each variant. The experiment between D0 and D5 was used for the adjustment of the position. Variants with under-represented read counts (<10) at D0 and D5 were excluded from further analysis. The log2 ratios were scaled within each exon by replicating the experiment. The average score for each variant was calculated from all of the non-missing values. Similar to the within exon normalizing, linear scaling was used to get scores across exons. After completion of all data cleaning and quality control, a raw functional score was available for 6,959 SNVs (Supplementary Table 3).
BRCA2 functionally PStrong missense alterations were mapped in the DBD using PyMol software. The NCBI Molecular Modeling Database has aProtein Data Bank source file. Three-dimensional structural modelling was based on the crystal structure of a BRCA2–DSS1–ssDNA complex39.
Align-GVGD was the place where the BRCA2 exonucing sequence was obtained. Sequence alignments were performed using ten species: Homo sapiens, Pan troglodytes, Macaca mulatta, Rattus norvegicus, Canis familiaris, Bos taurus, Monodelphis domestica, Gallus gallus, Xenopus laevis and Tetraodon nigroviridis. Sequence conservation analyses were performed on amino-acid residues that contained BRCA2 DBD functionally pathogenic variants. Align-GVGD26, AlphaMissense27 and Bayes-Del40 were used for in silico pathogenicity prediction.
The functional results from SGE were compared with the results of other studies, including the cell line–based drug assays and the mouse embryonic-stem-cell functional analysis.
The ACMG–AMP rule-based framework combines evidence from population, computational and predictive, segregation, functional, and other data, with each contributing source weighted as very strong (PVS1), strong (PS1, PS2, PS3 and PS4), moderate (PM1, PM2, PM3, PM4, PM5 and PM6) or supporting (PP1, PP2, PP3, PP4 and PP5) evidence for pathogenic effects, or stand-alone (BA1), strong (BS1, BS2, BS3 and BS4) or supporting (BP1, BP2, BP3, BP4, BP5, BP6 and BP7) for benign effects. Variant classifications are produced by combining the data for LB, pathogenic,LP and VUS9. ACMG–AMP scoring rules developed by the Clin Gen were used in this study for clinical classification of the BRCA2 SNVs. The BRCA2 data is integrated into the classification model under the PS3/BS3 rule. The values for functional evidence were capped at +4 and -4 on the scale, which made it difficult to get classification with functional evidence alone. The Western Institutional Review Board approved the study, as well as the clinical testing cohort and the Mayo Clinic Institutional Review Board. The Supplementary Methods contain detailed ACMG–AMP criteria used in the study.
LOH status for breast, ovarian, pancreatic, and prostate cancer tumours carrying germline BRCA2 DBD variants was acquired from tumour–normal paired sequencing using the IMPACT dataset32. The FACETS algorithm41 was used to determine LOH from matched tumour–normal pairs. The analysis only looked at tumour samples with over 40% content.
Optimal sgRNA selection for HAP1 cell transfection in Hoechst 34580 (BD, 5551877) and haploidy sorting with Integrated DNA Technologies
The data in this paper are provided with explicit written permission of the study participants after they have been approved by the institutional review boards.
HAP1cells were kept in theIMSM with 10% FBS and 1% penicillin–streptomycin. For haploidy sorting, 1 × 10−7 HAP1 cells were resuspended in 5 mg ml–1 Hoechst 34580 (BD, 565877) and sorted at 4 °C. The HAP1 cells were transfected using a method called Origene. All oligonucleotides and primers were synthesized by Integrated DNA Technologies.
Multiple sgRNAs with predicted high editing efficiency in HAP1 cells were evaluated in SGE experiments and the optimal sgRNAs were selected. Each experiment, 5 million haploid-sorted HAP1 cells were co-transfected with various amounts of the target-specific variant library as well as a small quantity of the sgRNA targeting construct. Puromycin was used to select cells for 3 days. Cells were collected at D0, D5 (24 h after puromycin selection) and D14 after transfection, and gDNA was extracted using a Monarch Genomic DNA Purification kit (New England Biolabs, T3010L). Target regions were amplified by PCR to add barcodes for multiplexing. All 50 l reactions were performed using the Q5 High-Fidelity 2 master mix. Primers for gDNA amplification are provided in Supplementary Table 2. All reactions were cleaned and concentrated using Ampure XP beads before sequencing for 150 cycles on an Illumina MiSeq (approximately 5 million reads per run) or NextSeq (approximately 30 million reads per run) instrument. In order to further process base calls, they used a custom program that was implemented using the instrument control software.


